Dashboard An overview of available bioinformatic tools.
CAT allows you to easily compare your peak-called ChIP peaks with those from most all D. melanogaster , mouse, and human modENCODE/ENCODE ChIP peaks.
Select the appropriate ChIP Association Tester to use
Chip Association of Drosophila proteins (dm3)
Chip Association of Mouse proteins (mm9)
Chip Association of Human proteins (hg19)
Chip Association of other data (not ENCODE)
Comparison is performed using the Genomic Association Tester that has been used to pre-compute the association of all proteins on chromatin used by ENCODE and modENCODE in their ChIP datasets.
Peaks that share significantly common genomic binding sites score highly, allowing easy identification of proteins that show similar or identical genomic distributions from those that are distinct.
How are ChIP association scores calculated?
Scores are calculated as:
Log2( observed overlap / expected overlap ) x -Log10( p-value ) x sensitivity coefficient
The sensitivity coefficient is what distinguishes overlapping ChIP peaks from highly similar ChIP peaks and is determined from the percent of total bases overlapping between the two ChIP tracks being tested.
If 100% of all bases in ChIP A overlap with 100% of all bases in ChIP B, then sensitivity coefficient will be 4 : log10(% overlap total A bases) x log10(% overlap total B bases) )
For example, if we take the following four ChIP peak BED files, ChIP A, ChIP B, ChIP C, and ChIP D :
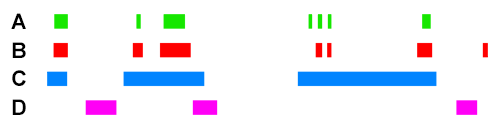
Scoring for overlapping ChIP peaks will cluster ChIPs A,B,C since they all overlap highly between themselves. This is likely the case for histone modifying enzymes that deposit broad peak histone marks.
Scoring for highly similar ChIP peaks will cluster ChIPs A and B since they both colocalise well together (>90% total bases of each overlapping). This would be the case for e.g. colocalising transcription co-factors.
ChIP C will still have an association score higher with A+B than ChIP D will have with A+B, because the observed overlap of C with A/B is still high. However, since the percent total bases of C is low (e.g. <10%), the sensitivity coefficient will lower the association score relative to the observed between A+B when scoring for highly similar peaks.
High genomic association or "co-localisation" between pairs of ChIP data can highlight potential :
- functional complexes or co-factors acting on similar genes.
- antagonistic activators/repressors acting on similar genes.
- preference for a given chromatin-acting protein with one or more histone marks.
- co-occurring histone marks
-
Uploading your own BED file will allow comparison of association of modENCODE ChIP peaks with peaks/genomic regions of your supplied BED file.
- This takes approximately 10 minutes for comparison of 1000 ChIP peaks against 200 other factors, depending on the current server load.
-
The supplied BED file is usually a ChIP peak BED file which will compare "all ChIP peaks with all ChIP peaks".
However, you may wish to test association of modENCODE ChIP peaks with individual genomic regions (e.g. genes).- If this is the case, you should check the option "Individual Genomic Regions", which will compare all the selected modENCODE ChIP peaks with each individual genomic region in the provided BED file.
- Due to the time involved in calculating peak association, uploading your own ChIP peak BED file requires the input of a valid email address to which the results will be sent.
- User-supplied ChIP peak BED files are limited to 5 ChIP peak BED files (unlimited regions), although if you require individual region testing (e.g. a gene BED file), then only 1 BED file is permitted, with a maximum of regions.
